
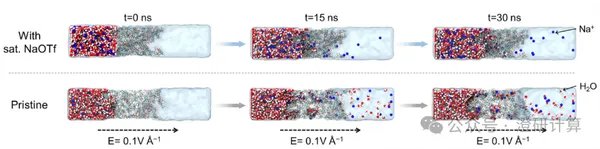
Ion Diffusion / Battery / Porous materials and separation transport
Case study from Nature Commun and Angewandte Chemie: How to use molecular dynamics (MD) to quantitatively analyze ion/water migration in membrane diffusion?
Read OriginalIn battery development, experimental testing can tell us about macroscopic polarization and rate performance; however, how ions move within the membrane pores/interfaces often requires molecular dynamics (MD) to "see and explain."
In battery membrane research, MD primarily undertakes three core tasks:
1. Visualizing transport paths: Visualizing the abstract "ion channels," intuitively showing whether ions creep along polymer chains or diffuse freely in water-filled channels.
2. Quantifying selectivity mechanisms: Calculating the migration rate and flux ratio of different ions in the membrane through non-equilibrium (NEMD) simulations of applied external fields, directly quantifying membrane selectivity.
3. Analyzing solvation effects: Accurately calculating the coordination number of ions, revealing the energy cost of "desolvation" or "water-carrying migration."
Read OriginalIn battery membrane research, MD primarily undertakes three core tasks:
1. Visualizing transport paths: Visualizing the abstract "ion channels," intuitively showing whether ions creep along polymer chains or diffuse freely in water-filled channels.
2. Quantifying selectivity mechanisms: Calculating the migration rate and flux ratio of different ions in the membrane through non-equilibrium (NEMD) simulations of applied external fields, directly quantifying membrane selectivity.
3. Analyzing solvation effects: Accurately calculating the coordination number of ions, revealing the energy cost of "desolvation" or "water-carrying migration."
![[Coating MD] From Micropores to Hierarchical Pores: What exactly should battery coating MD simulation be considered?](https://img.chengyankeji.cn/uploads/optimized/1776753751718-20260313-w600.webp)
Porous materials and separation transport / Ion Diffusion
[Coating MD] From Micropores to Hierarchical Pores: What exactly should battery coating MD simulation be considered?
Read OriginalIn battery research, the construction of coatings, such as artificial solid electrolyte interfaces (ASEI) or porous framework coatings on electrode surfaces, is a popular research topic.
To thoroughly explain the mechanism of action of coatings in an article, molecular dynamics (MD) simulations are an indispensable tool. It is worth noting that the focus of MD calculations differs depending on the pore size. This article, based on five high-level papers, extracts the MD calculations of coatings into two core physical dimensions according to the material's pore size:
● Microporous systems (pore size < 1 nm): Focusing on desolvation barriers and the "dynamic shuttle" of ions within the coating.
● Mesoporous and hierarchical pore systems (pore size > 2 nm): Focusing on local solvation structures, long-range diffusion behavior, and the synergistic effect of hierarchical channels.
Read OriginalTo thoroughly explain the mechanism of action of coatings in an article, molecular dynamics (MD) simulations are an indispensable tool. It is worth noting that the focus of MD calculations differs depending on the pore size. This article, based on five high-level papers, extracts the MD calculations of coatings into two core physical dimensions according to the material's pore size:
● Microporous systems (pore size < 1 nm): Focusing on desolvation barriers and the "dynamic shuttle" of ions within the coating.
● Mesoporous and hierarchical pore systems (pore size > 2 nm): Focusing on local solvation structures, long-range diffusion behavior, and the synergistic effect of hierarchical channels.
![[Diffusion within pores] Narrower pores, faster diffusion? Molecular dynamics reveals a "super high-speed rail" diffusion mechanism in nanopores. (Zheng Anmin, Nature Communications)](https://img.chengyankeji.cn/uploads/optimized/1776756402547-20260324-w600.webp)
Porous materials and separation transport
[Diffusion within pores] Narrower pores, faster diffusion? Molecular dynamics reveals a "super high-speed rail" diffusion mechanism in nanopores. (Zheng Anmin, Nature Communications)
Read OriginalIn the study of porous materials, especially molecular sieves, diffusion rate often directly determines material properties. Our intuition is usually that the narrower the pores, the stronger the confinement, and the slower the molecular diffusion should be.
However, this work by Zheng Anmin's team, published in Nature Communications, overturns this understanding: for long-chain molecules, smaller pores actually lead to faster diffusion.
Read OriginalHowever, this work by Zheng Anmin's team, published in Nature Communications, overturns this understanding: for long-chain molecules, smaller pores actually lead to faster diffusion.
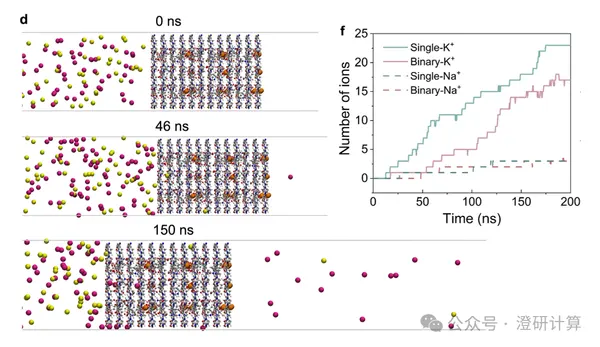
Porous materials and separation transport / Ion Diffusion
COF can actually perform these calculations? Nat. Commun. uses DFT and molecular dynamics to explain the K+/Na+ separation mechanism.
Read OriginalThe 2025 Nobel Prize in Chemistry was awarded to MOFs, bringing greater attention to framework materials like MOFs and COFs. What theoretical calculations can be performed on MOF/COF materials?
Read Original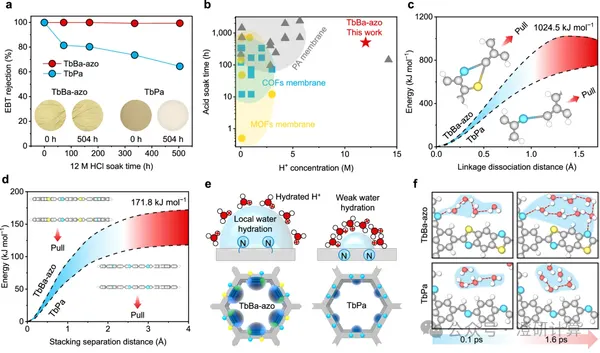
Ion Diffusion / Porous materials and separation transport
A COF membrane paper, with theoretical calculations making up almost half of it? This Nat. Commun. paper thoroughly explains the mechanism using DFT, MD, AIMD, and PMF.
Read OriginalFor many materials science articles to be published in top journals, performance results alone are no longer sufficient. This is especially true when working on membrane and interface materials; explaining the mechanisms through theoretical calculations has become an almost indispensable step. Experiments can tell you that "the results have improved," but if you want to further explain "why they have improved, what the structural changes have actually brought about, and whether this mechanism is credible," you often need to rely on theoretical calculations to complete the logical chain.
Read Original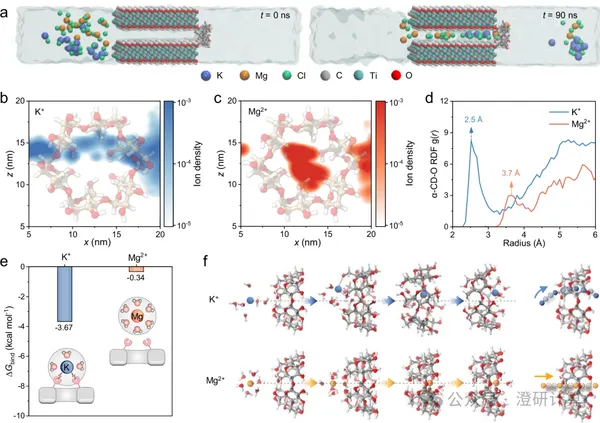
Porous materials and separation transport
Interpretation of Nat. Commun. Reviewer Responses: Supplement and Explanation of DFT and MD Models in MXene Ion Separation Membranes
Read OriginalIn peer review responses, theoretical calculations are not only used to explain experimental results, but also often require further explanation of the model configuration, the objects of calculation, and the physical meaning of the results. This Nature Communications work immobilized α-cyclodextrin at the entrance of the MXene interlayer channel and used DFT and MD to explain K+/Mg2+ separation. Reviewers raised several comments regarding hydrated ion binding, α-CD adsorption configuration, the rationality of the MD model, and the selection of calculated ions. The authors responded to each point by supplementing the adsorption configuration, concentration experiments, and model comparisons.
Read Original