
Battery
Nature Energy: How to make changing a solvent molecule sound sophisticated? From DFT, MD to CDFT, see how theoretical calculations can elevate the quality of your papers.
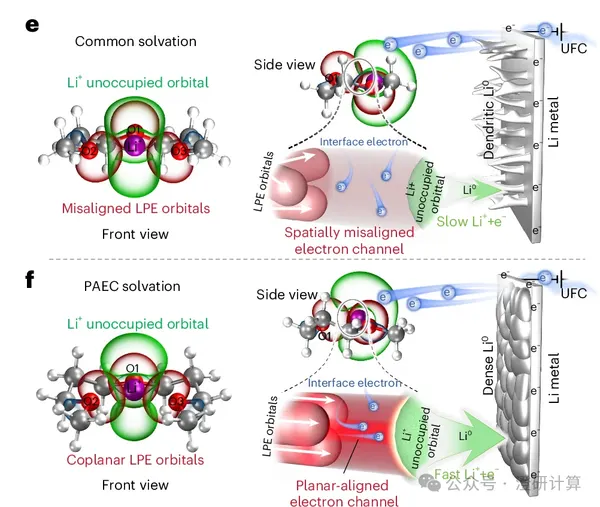
Read OriginalMany electrolyte research papers, on the surface, seem to focus on just one thing: changing a solvent molecule.
However, those that truly get published in high-level journals are rarely as simple as "I changed the molecule, so the performance is better." Rather, they demonstrate whether the authors can elevate this molecular change to a new structural concept, a new interface mechanism, and a complete, verifiable chain of theoretical calculations.
Read OriginalHowever, those that truly get published in high-level journals are rarely as simple as "I changed the molecule, so the performance is better." Rather, they demonstrate whether the authors can elevate this molecular change to a new structural concept, a new interface mechanism, and a complete, verifiable chain of theoretical calculations.
Battery
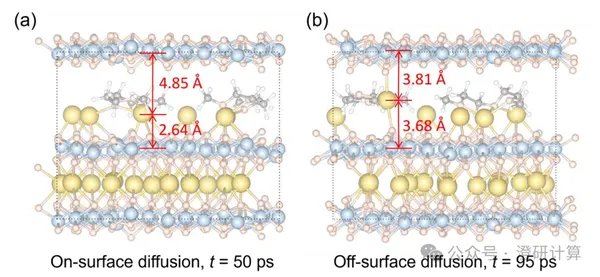
Nature Energy: Can electrolyte solvents also reversibly embed into layered cathodes? DFT, CI-NEB, and AIMD explain a new mechanism for ultra-fast charging sodium batteries.
Read OriginalIn traditional sodium-ion batteries, it is generally assumed that only Na⁺ undergoes reversible insertion/extraction within the layered cathode, while the electrolyte solvent is only responsible for coordination and transport in the liquid phase. This work, published in Nature Energy by Qi Liu's team at City University of Hong Kong, proposes a different reaction scenario: in a diglyme electrolyte, solvent molecules can reversibly insert into the layered manganese-based cathode under high voltage, altering the interlayer diffusion environment and thus enhancing Na⁺ migration and bulk redox kinetics under ultrafast charging conditions.
Read Original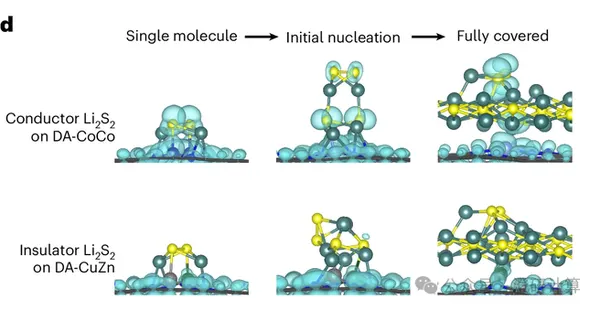
Catalysis / Battery
Nature Catalysis: New Criteria for Lithium-Sulfur Catalysis: Theoretical Calculations Reveal that Energy Barriers Cannot Be the Only Factor in Lithium-Sulfur Reactions
Read OriginalIn the past, when working with lithium-sulfur and lithium-oxygen systems, people were more accustomed to using thermodynamic indicators such as adsorption energy, reaction energy barrier, and free energy to screen catalysts. However, what truly blocks the reaction is often not whether the first step of the reaction can occur, but rather that the accumulation of insulating solid intermediates such as Li₂S₂ and Li₂O₂ blocks electron transport, making it increasingly difficult for subsequent reactions to continue.
Read Original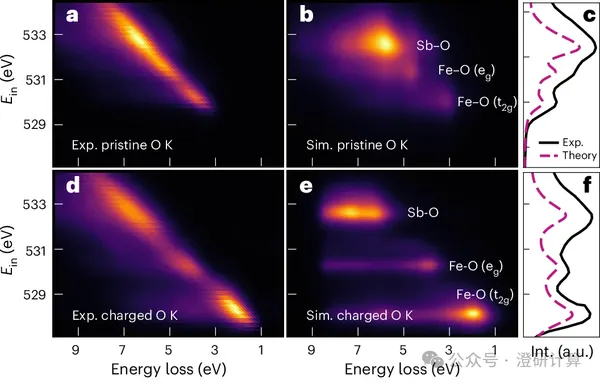
Battery
Nature Materials: Iron-based cathode achieves high-valence FeIII-V redox; theoretical calculations reveal 3d⁵L² negative charge transfer state
Read OriginalIron-based cathode materials are inexpensive and abundant, but the commonly used FeII/III redox potentials are limited. To further increase the voltage, it is necessary to move to the redox potentials of higher valence iron (FeIII and above); the difficulty lies in the fact that higher valence Fe is usually unstable and easily accompanied by oxygen oxidation, O–O dimerization, and voltage hysteresis.
Read Original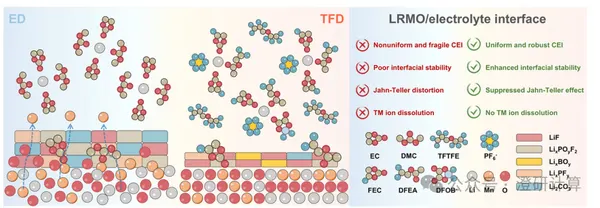
Battery
Gangcheng Da Liuqi AM: What calculations can be performed by regulating the CEI of the positive electrode with electrolyte?
Read OriginalElectrolyte-mediated regulation of the cathode/CEI can be calculated along two lines: first, where does the CEI come from and can it protect the cathode surface; second, whether electrolyte molecules will further affect the Mn–O local structure and Jahn–Teller distortion.
Read Original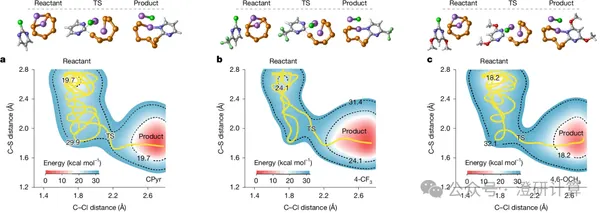
Battery
Latest Nature report on lithium-sulfur batteries! Quantum chemistry + machine learning design of lithium-sulfur precursors.
Read OriginalMolecular mediators in lithium-sulfur batteries are essentially a class of organic molecular additives added to the electrolyte. They interact with polysulfides to alter sulfur conversion pathways, reduce reaction polarization, and improve the Li₂S deposition process. Past molecular additive design focused more on functional groups, such as their ability to bind polysulfides or promote Li₂S formation. This Nature paper by Zhou Guangmin's team at Tsinghua University's Shenzhen International Graduate School focuses on the molecular skeleton of organic additives: even within the CPyr class of molecules, different substituents and substitution sites simultaneously affect the rate at which the molecule is activated by polysulfides, as well as the charge transfer capability of the activated mediator.
Read Original