
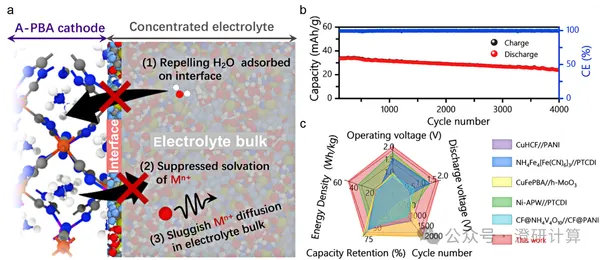
Battery
Interface distribution, free water, and diffusion coefficient: how molecular dynamics (MD) explains the dissolution failure of aqueous electrodes in three steps?
Read OriginalIn aqueous batteries, Prussian blue and its analogues (PBA/PBAs) are very attractive: open structure, rapid diffusion, and low cost. However, in actual battery applications, many systems exhibit significant capacity degradation within a few dozen cycles. One common reason is the dissolution of transition metals: once metal ions like Mn/Fe begin to enter the electrolyte, the framework gradually becomes hollow/defective, causing a simultaneous drop in capacity and voltage plateau.
This 2022 ACS Energy Letters work may not be "new" in itself, but it demonstrates how to use theoretical calculations to break down experimental phenomena into essential microscopic dynamic steps and clearly reveal the mechanism using quantifiable indicators.
Read OriginalThis 2022 ACS Energy Letters work may not be "new" in itself, but it demonstrates how to use theoretical calculations to break down experimental phenomena into essential microscopic dynamic steps and clearly reveal the mechanism using quantifiable indicators.
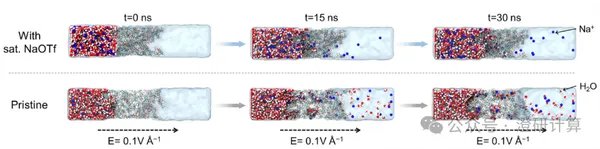
Ion Diffusion / Battery / Porous materials and separation transport
Case study from Nature Commun and Angewandte Chemie: How to use molecular dynamics (MD) to quantitatively analyze ion/water migration in membrane diffusion?
Read OriginalIn battery development, experimental testing can tell us about macroscopic polarization and rate performance; however, how ions move within the membrane pores/interfaces often requires molecular dynamics (MD) to "see and explain."
In battery membrane research, MD primarily undertakes three core tasks:
1. Visualizing transport paths: Visualizing the abstract "ion channels," intuitively showing whether ions creep along polymer chains or diffuse freely in water-filled channels.
2. Quantifying selectivity mechanisms: Calculating the migration rate and flux ratio of different ions in the membrane through non-equilibrium (NEMD) simulations of applied external fields, directly quantifying membrane selectivity.
3. Analyzing solvation effects: Accurately calculating the coordination number of ions, revealing the energy cost of "desolvation" or "water-carrying migration."
Read OriginalIn battery membrane research, MD primarily undertakes three core tasks:
1. Visualizing transport paths: Visualizing the abstract "ion channels," intuitively showing whether ions creep along polymer chains or diffuse freely in water-filled channels.
2. Quantifying selectivity mechanisms: Calculating the migration rate and flux ratio of different ions in the membrane through non-equilibrium (NEMD) simulations of applied external fields, directly quantifying membrane selectivity.
3. Analyzing solvation effects: Accurately calculating the coordination number of ions, revealing the energy cost of "desolvation" or "water-carrying migration."
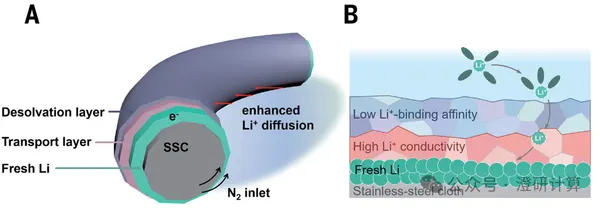
Battery
Science Computation Intensive Reading: How to Use Multi-Scale Computation CMD, AIMD, ReaxFF, and COMSO to Completely Understand the Multi-Scale Mechanism of the SEI Interface?
Read OriginalThe solid electrolyte interface (SEI) plays a crucial role in both battery storage and electrocatalysis systems. However, experimental characterization often only provides macroscopic electrochemical performance, making it difficult to directly observe the dynamic evolution of ions at the interface. If we want to thoroughly explain the mechanism of the SEI in a top-tier journal, what can theoretical calculations do?
Read Original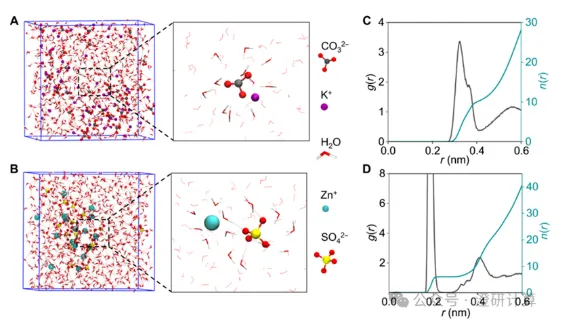
Popular Science
Why is calculating the electrolyte molecular weight (MD) so expensive? What data do I need to prepare before hiring someone to do it? What results can I expect?
Read OriginalAnyone who has used agencies to perform electrolyte kinetic simulations knows that calculating the molecular dynamics (MD) of an electrolyte typically costs several times more than calculating binding energy, electrostatic potential, adsorption energy, and other parameters.
Many people think that MD simulation is simply running a testing software program—you input molecules, click "Run," and get the graph. However, in reality, there's a lot of preparatory work required before "Running." How much work is involved, what data needs to be prepared before hiring someone to perform the calculation, and what results can you get afterward? This article will provide a detailed explanation.
Read OriginalMany people think that MD simulation is simply running a testing software program—you input molecules, click "Run," and get the graph. However, in reality, there's a lot of preparatory work required before "Running." How much work is involved, what data needs to be prepared before hiring someone to perform the calculation, and what results can you get afterward? This article will provide a detailed explanation.
![[CEI Calculation] Wang Chunsheng's In-depth Reading of Nat. Chem. Calculation - How exactly is the positive electrode interface membrane (CEI) calculated?](https://img.chengyankeji.cn/uploads/optimized/1776753566297-20260312-w600.webp)
Battery
[CEI Calculation] Wang Chunsheng's In-depth Reading of Nat. Chem. Calculation - How exactly is the positive electrode interface membrane (CEI) calculated?
Read OriginalIn battery development, the SEI (solid electrolyte interphase) of the negative electrode has been studied relatively thoroughly. In contrast, the CEI of the positive electrode is much more complex. If we want to fully explain the dynamic mechanism of CEI in top journals, what can theoretical calculations do? Today, we will combine two papers, namely Professor Chunsheng Wang's team from the University of Maryland in *Nature Chemistry* and Chih-Chiang Chiang's team from National Taiwan University in *J. Chem. Phys.*, to interpret what theoretical calculations can be performed on CEI.
Read Original![[Coating MD] From Micropores to Hierarchical Pores: What exactly should battery coating MD simulation be considered?](https://img.chengyankeji.cn/uploads/optimized/1776753751718-20260313-w600.webp)
Porous materials and separation transport / Ion Diffusion
[Coating MD] From Micropores to Hierarchical Pores: What exactly should battery coating MD simulation be considered?
Read OriginalIn battery research, the construction of coatings, such as artificial solid electrolyte interfaces (ASEI) or porous framework coatings on electrode surfaces, is a popular research topic.
To thoroughly explain the mechanism of action of coatings in an article, molecular dynamics (MD) simulations are an indispensable tool. It is worth noting that the focus of MD calculations differs depending on the pore size. This article, based on five high-level papers, extracts the MD calculations of coatings into two core physical dimensions according to the material's pore size:
● Microporous systems (pore size < 1 nm): Focusing on desolvation barriers and the "dynamic shuttle" of ions within the coating.
● Mesoporous and hierarchical pore systems (pore size > 2 nm): Focusing on local solvation structures, long-range diffusion behavior, and the synergistic effect of hierarchical channels.
Read OriginalTo thoroughly explain the mechanism of action of coatings in an article, molecular dynamics (MD) simulations are an indispensable tool. It is worth noting that the focus of MD calculations differs depending on the pore size. This article, based on five high-level papers, extracts the MD calculations of coatings into two core physical dimensions according to the material's pore size:
● Microporous systems (pore size < 1 nm): Focusing on desolvation barriers and the "dynamic shuttle" of ions within the coating.
● Mesoporous and hierarchical pore systems (pore size > 2 nm): Focusing on local solvation structures, long-range diffusion behavior, and the synergistic effect of hierarchical channels.